
OP23 Efficacy and safety of vedolizumab SC in patients with moderately to severely active Crohn’s disease: Results of the VISIBLE 2 study
S. Vermeire1, W. Sandborn2, F. Baert3, S. Danese4, T. Kobayashi5, E.V. Loftus Jr6, S. Bhatia7, K. Kisfalvi8, M. Rosario9, W. Zhang10, G. D’Haens11
1Department of Gastroenterology, Leuven, Belgium, 2Division of Gastroenterology, La Jolla, University of California - San Diego, USA, 3Department of Gastroenterology, AZ Delta, Roeselare, Belgium, 4Gastrointestinal Immunopathology, Humanitas University, Milan, Italy, 5Center for Advanced IBD Research and Treatment, Kitasato University, Tokyo, Japan, 6Division of Gastroenterology and Hepatology, Mayo Clinic College of Medicine, Rochester, USA, 7Takeda, Medical Safety, London, UK, 8Takeda, Research and Development- Global Clinical Sciences, Cambridge, USA, 9Takeda, Clinical Pharmacology, Cambridge, USA, 10Takeda, Statistical and Quantitative Sciences, R&D, Cambridge, USA, 11Department of Gastroenterology, Amsterdam University Medical Centers, Amsterdam, The Netherlands
Background
Vedolizumab (VDZ) is a gut-selective, humanised, monoclonal α 4β 7 integrin antibody for the treatment of patients with moderately to severely active ulcerative colitis (UC) or Crohn’s disease (CD). VDZ is currently an intravenous (IV) therapy; a subcutaneous (SC) formulation is under development to provide patients with an alternative route of administration for maintenance treatment for UC and CD. Here we present the first data from the phase 3 study of VDZ SC maintenance treatment in CD.
Methods
VISIBLE 2 (NCT02611817; EudraCT 2015-000481-58) was a randomised, double-blind, placebo (PBO)-controlled phase 3 trial of VDZ SC as maintenance treatment in adults with moderately to severely active CD. Patients (
Results
Patients who responded to VDZ IV induction at Week 6 (
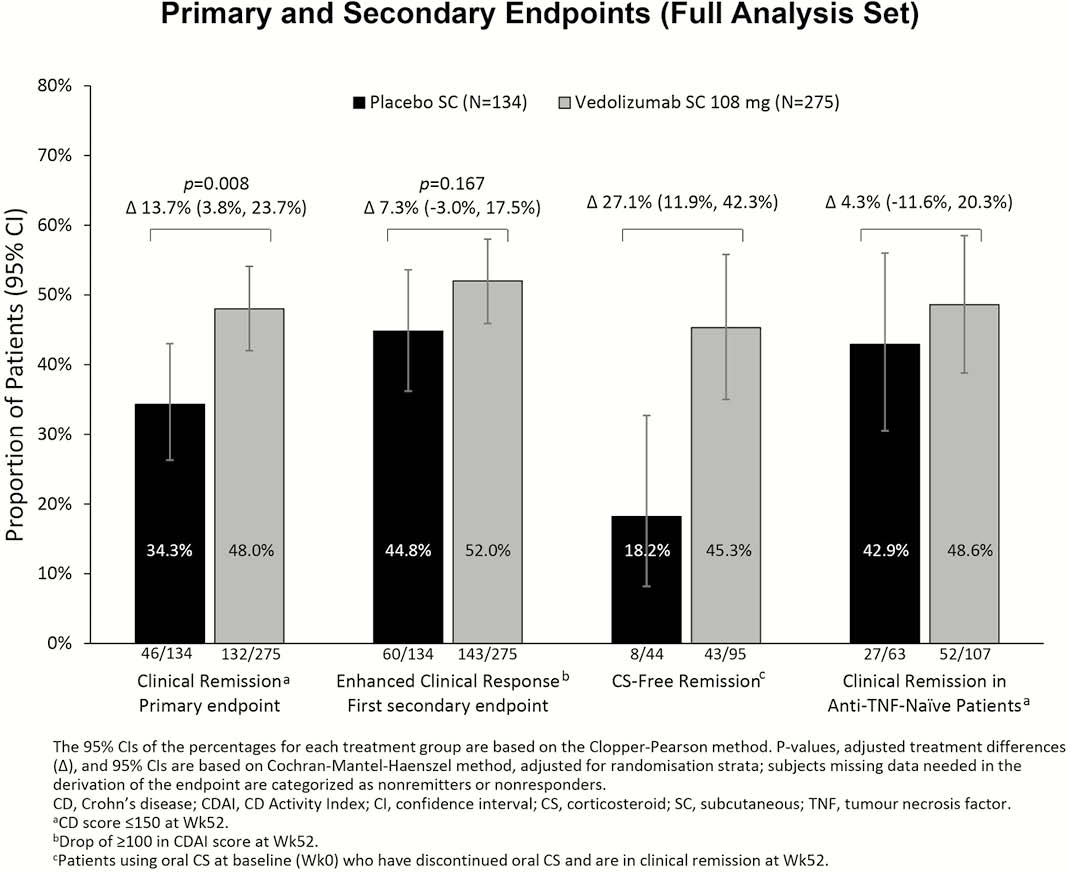
Conclusion
Among VDZ IV induction responders, significantly more patients on maintenance VDZ SC than PBO achieved clinical remission at Week 52. The safety findings with VDZ SC remain in line with the known safety profile of VDZ IV in patients with CD.