
OP30 The interplay of microbiome dysbiosis and immune system deregulation in patients with Crohn’s disease
N.S. Seyed Tabib1, C. Caenepeel1, K. Machiels1, S. Verstockt1, B. Verstockt1,2, N. Ardeshir Davani1, J. Sabino1,2, M. Ferrante1,2, S. Vermeire1,2
1Chronic Diseases, Metabolism and Ageing, KU Leuven, Leuven, Belgium, 2Department of Gastroenterology and Hepatology, University Hospitals Leuven, Leuven, Belgium
Background
The perturbation of composition, function, and structure of the gut microbiota known as dysbiosis is a key factor in inflammatory bowel disease (IBD) pathogenesis. There is a crosstalk between the microbiota and the gut immunological niche. To better understand this interaction, we characterised the degree of dysbiosis and dysregulation of the immune proteome in Crohn’s disease (CD) patients to see whether subtypes of patients could be identified.
Methods
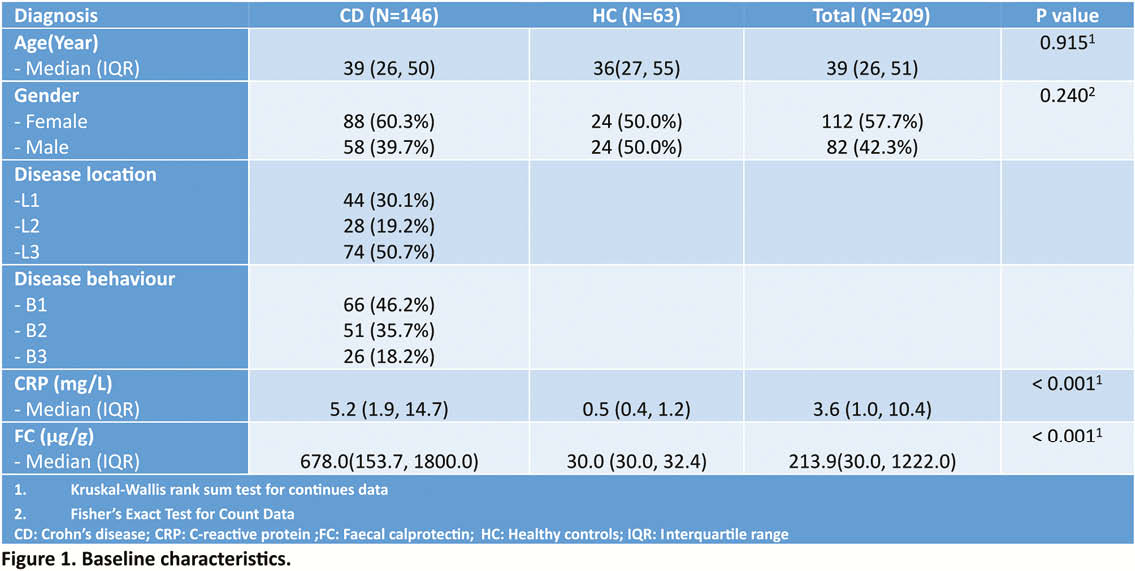
We collected faecal and serum samples of 146 CD patients and 63 healthy controls (HC) (Figure 1), and studied microbiota phylogenetic (16S rRNA gene sequencing) and serum proteomic (91 inflammatory proteins OLINK). Microbial dysbiotic index (MDI), defined as the logarithm of the sum of [abundance in organisms increased in CD] over the [abundance of organisms decreased in CD] was calculated and patients were ranked from Q1 (the least dysbiotic state) to Q4 (the most dysbiotic state). For the proteomic score, 32 proteins that correlated (adj.
Results
The MDI did not correlate with standard phenotypic subgroups based on the Montreal classification but did positively correlate with C-reactive protein (CRP) and FC level (
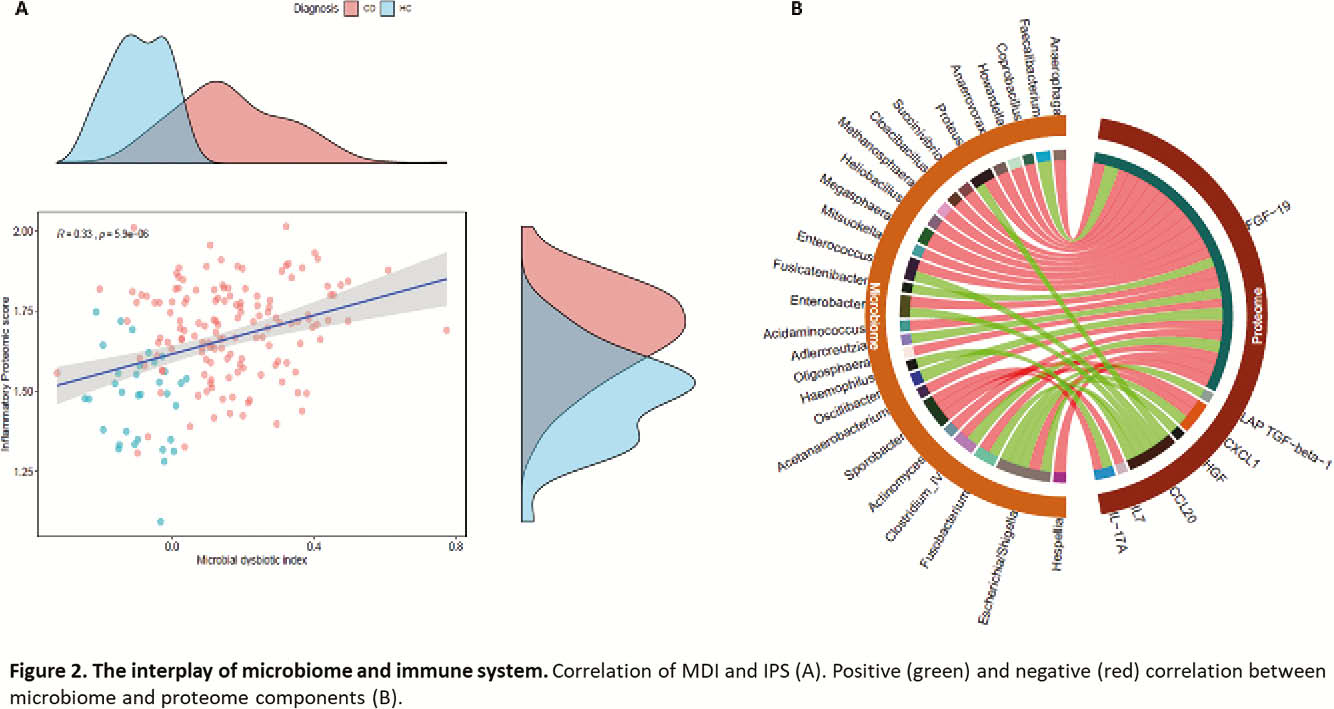
Conclusion
We were able to define clusters of patients based on molecular characterisation of different players in IBD pathogenesis such as microbiota and proteome. This molecular clustering in a given patient could be considered as a novel therapeutic and personalised approach to IBD. Further validation in larger cohorts is required.