
P680 Similar gut bacterial composition between patients with ulcerative colitis and healthy controls in a high prevalence population: a cross-sectional study of the Faroe Islands IBD cohort
Á Fríðriksmørk BerbisáCand.Scient, M.(1);Rubek Nielsen, K.(2);Midjord, J.(2);Oddmarsdóttir Gregersen , N.(3);Ingham, A.C.(4);Burisch, J.(5);Gratton Vang, A.(1);
(1)Fróðskaparsetur Føroya, Department of Health Science, Tórshavn, Faroe Islands;(2)National Hospital of the Faroe Islands, Department of Medicine, Tórshavn, Faroe Islands;(3)Genetic Biobank of the Faroe Islands, FarGen, Tórshavn, Faroe Islands;(4)Statens Serum Institut, Dept. of Bacteria- Parasites and Fungi, Copenhagen, Denmark;(5)North Zealand University Hospital, Department of Gastroenterology, Fredrikssund, Denmark
Background
The Faroe Islands, a genetically and geographically isolated population in the North Atlantic, has the world’s highest prevalence of inflammatory bowel disease (IBD). Epidemiological studies have characterized this unique cohort, which consists predominately of an ulcerative colitis phenotype, and a decreased risk of developing IBD with emigration. Therefore, the well-characterized Faroese IBD cohort gives the opportunity to better understand the interplay between genetic predisposition and environmental triggers for this complex disease. This study represents the first investigation into the composition of the gut microbiome for the cohort.
Methods
This cross-sectional study consisted of 41 patients with established ulcerative colitis (UC) and 144 age-and sex matched healthy controls (HC) recruited through the Faroese Genome project (FarGen). Participants donated a one-time fecal sample and completed questionnaires on food-frequency, background health and lifestyle. DNA extractions were completed and 16S rRNA amplicon sequencing of the V3-V4 region was performed followed by bioinformatic analysis of taxonomy and diversity metrics.
Results
The overall composition of the sequenced fecal bacteria was dominated by Firmicutes (64,5% of total amplicon sequence variants) and Bacteroidetes (20,95% of total amplicon sequence variants). Akkermansia muciniphila was absent from 30% of the study samples. Discriminatory analysis for indicator taxa identified that Coprococcus and Prevotella_9 characterized healthy controls while genus Ruminiclostridium characterizes UC patients. However, no statistically significant differences were found between UC and HC based on metrics of alpha or beta diversity. Food frequency questionnaires revealed no differences in dietary patterns of the two groups.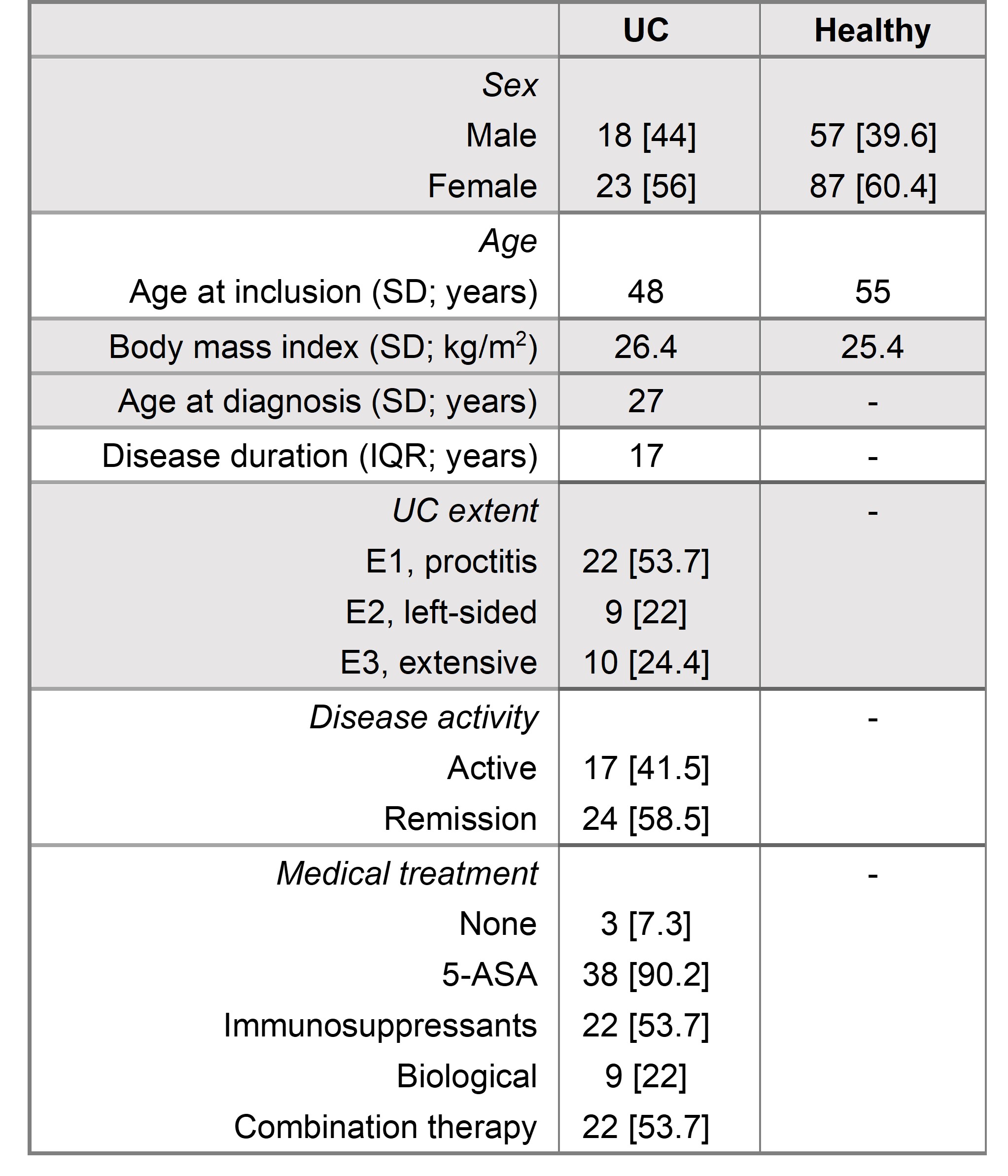
Table 1 Data is expressed as number [%], median [IQR] or mean [SD]. The study cohort included 185 participants with 41 patients diagnosed with ulcerative colitis and 144 healthy controls.

Figure 1 Heat trees showing grouped analysis with summed relative abundance of taxa for a) healthy control participants and b) Ulcerative colitis patients.
Conclusion
The similarity between the microbiome of Faroese UC and HC samples within this study and the absence of the beneficial taxa Akkermansia muciniphila in both groups, raises further questions concerning the underlying susceptibility toward inflammatory and metabolic disorders within this population. This study represents the first 16s rRNA amplicon sequencing data and insights into the Faroese UC and background population. The results highlight the importance of having regionally tuned reference ranges for microbial data.